06 Avr Des mutations de l’ADN non codant protègent le cerveau contre la SLA
Des chercheurs de l’Institut Weizmann des sciences découvrent une nouvelle voie neuroprotectrice.
Les mutations génétiques liées à une maladie sont souvent synonymes de mauvaises nouvelles. Des mutations dans plus de 25 gènes, par exemple, sont associées à la sclérose latérale amyotrophique, ou SLA, et elles augmentent toutes le risque de développer cette maladie incurable. Aujourd’hui, une équipe de recherche dirigée par le professeur Eran Hornstein, de l’Institut Weizmann des Sciences, a établi un lien entre un nouveau gène et la SLA, mais ce gène contient des mutations d’un type différent : elles semblent jouer un rôle défensif, plutôt qu’offensif, dans la maladie.
Le gène nouvellement lié à la SLA est situé dans la partie de notre génome autrefois appelée « ADN poubelle ». Cet ADN représente plus de 97 % du génome, mais comme il ne code pas de protéines, il était autrefois considéré comme de l’« ADN poubelle ». Aujourd’hui, bien que cet ADN non codant soit toujours considéré comme de la matière noire biologique, on sait déjà qu’il sert de manuel d’instructions crucial. Entre autres choses, il détermine quand les gènes de l’ADN codant – ceux qui codent les protéines – sont activés et désactivés.

(de gauche à droite) Prof. Eran Hornstein, Dr. Chen Eitan, and Aviad Siany
Le laboratoire du professeur Hornstein, au sein des Départements de Neurosciences Moléculaires et de Génétique Moléculaire du Weizmann, étudie les maladies neurodégénératives, c’est-à-dire les maladies dans lesquelles les neurones dégénèrent et meurent. L’équipe se concentre sur notre ADN non codant. « Cette partie massive et non codante du génome a été négligée dans la recherche des origines génétiques des maladies neurodégénératives comme la SLA », explique le professeur Hornstein. « Et ce, malgré le fait que pour la plupart des cas de SLA, les protéines ne peuvent pas expliquer l’émergence de la maladie ».
Beaucoup de gens connaissent la SLA grâce au défi du seau d’eau glacée qui est devenu viral il y a quelques années. Cette maladie neurologique rare s’attaque aux neurones moteurs, les cellules nerveuses responsables du contrôle des mouvements musculaires volontaires impliqués dans tout, de la marche à la respiration en passant par la parole. Les neurones meurent progressivement, entraînant finalement une insuffisance respiratoire et la mort. L’un des symptômes de la SLA est une inflammation dans les régions du cerveau liées aux neurones mourants, causée par des mécanismes immunitaires dans le cerveau.
« Notre cerveau possède un système immunitaire », explique le Dr Chen Eitan, qui a dirigé l’étude dans le laboratoire de Hornstein avec Aviad Siany. « Si vous êtes atteint d’une maladie dégénérative, les cellules immunitaires de votre cerveau, appelées microglies, vont essayer de vous protéger, en attaquant la cause de la neurodégénérescence. »
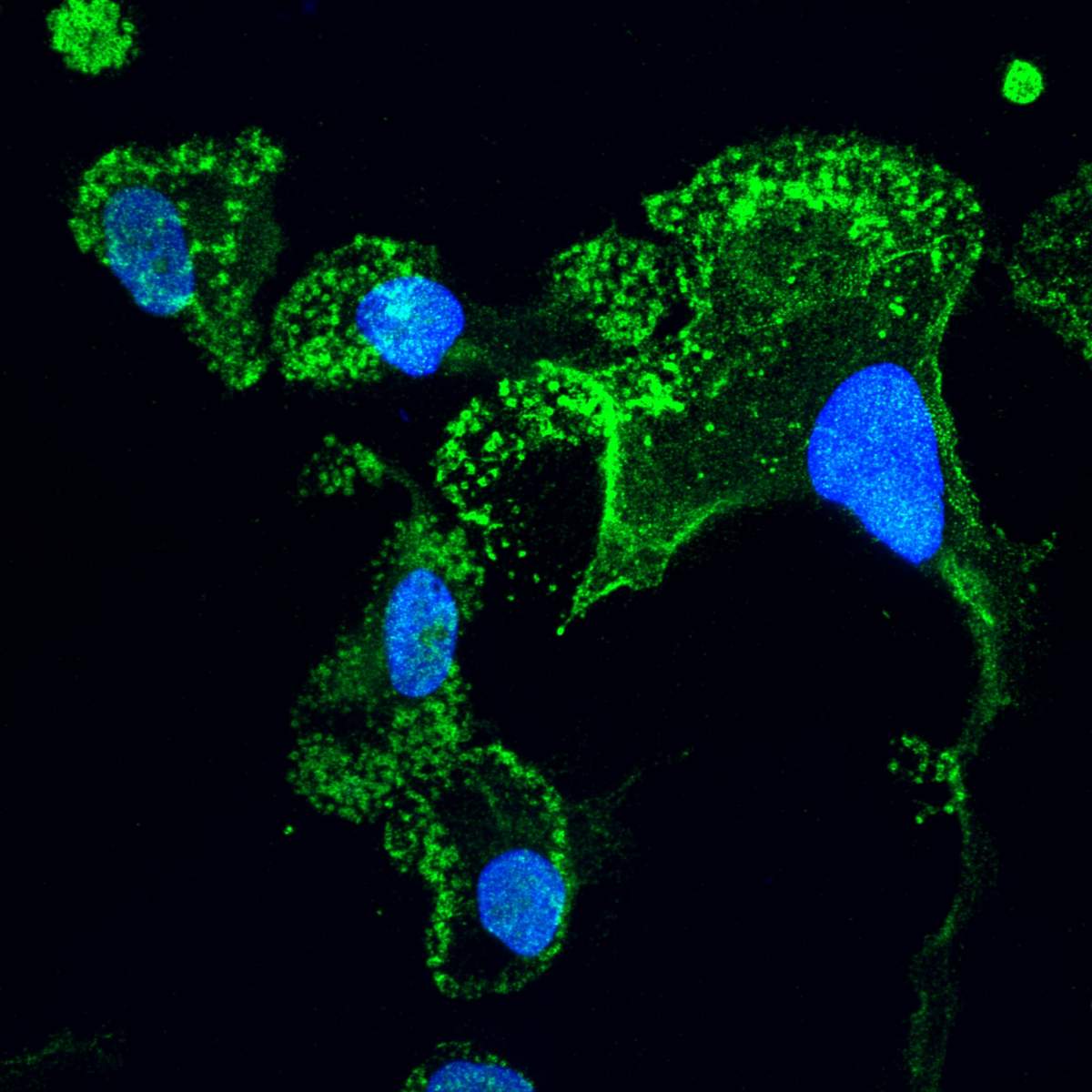
Microglie (en vert) qui a été « mûrie » en laboratoire à partir de cellules souches de patients atteints de SLA ; les noyaux des cellules sont en bleu. Vue en microscopie confocale
Le problème est que dans la SLA, la neurodégénérescence devient si grave que l’activation chronique de la microglie dans le cerveau atteint des niveaux extrêmement élevés, devenant toxique. Le système immunitaire finit donc par endommager le cerveau qu’il était censé protéger, ce qui entraîne la mort d’un plus grand nombre de motoneurones.
C’est là qu’interviennent les nouvelles découvertes, publiées aujourd’hui dans Nature Neuroscience. Les scientifiques de Weizmann se sont concentrés sur un gène appelé IL18RAP, connu depuis longtemps pour affecter la microglie, et ont découvert qu’il peut contenir des mutations qui atténuent les effets toxiques de la microglie. « Nous avons identifié des mutations dans ce gène qui réduisent l’inflammation », explique Eitan.
Après avoir analysé les génomes de plus de 6 000 patients atteints de SLA et de plus de 70 000 personnes qui n’en sont pas atteintes, les chercheurs ont conclu que les mutations nouvellement identifiées divisent par près de cinq le risque de développer la SLA. Il est donc extrêmement rare que les patients atteints de SLA présentent ces mutations protectrices, et les rares patients qui en sont porteurs ont tendance à développer la maladie environ six ans plus tard, en moyenne, que ceux qui ne présentent pas ces mutations. En d’autres termes, les mutations semblent être liées à un processus central de la SLA, ralentissant la maladie.
Pour confirmer ces résultats, les chercheurs ont utilisé une technologie d’édition de gènes pour introduire les mutations protectrices dans les cellules souches de patients atteints de SLA, ce qui a permis à ces cellules en culture de se transformer en microglies. Ils ont ensuite cultivé des microglies, avec ou sans les mutations protectrices, dans les mêmes boîtes avec des motoneurones. Les microglies portant les mutations protectrices se sont révélées moins agressives envers les motoneurones que les microglies dépourvues de ces mutations. « Les motoneurones ont survécu beaucoup plus longtemps lorsqu’ils étaient cultivés avec des microglies protectrices plutôt qu’avec des microglies normales », explique Siany.
Le Dr Chen Eitan note que ces résultats ont des implications potentielles pour la recherche sur la SLA et au-delà. « Nous avons découvert une nouvelle voie neuroprotectrice », dit-elle. « Des études futures pourront vérifier si la modulation de cette voie peut avoir un effet positif sur les patients. Sur un plan plus général, nos résultats indiquent que les scientifiques ne devraient pas ignorer les régions non codantes de l’ADN – non seulement dans la recherche sur la SLA, mais aussi dans l’étude d’autres maladies ayant une composante génétique. »
Les recherches du professeur Eran Hornstein sont soutenues par le Andrea L. and Lawrence A. Wolfe Family Center for Research on Neuroimmunology and Neuromodulation, le Kekst Family Institute for Medical Genetics, le Weizmann-Brazil Center for Neurodegeneration Research, le Nella and Leon Benoziyo Center for Neurological Diseases, la Goldhirsh-Yellin Foundation, la Redhill Foundation – Sam and Jean Rothberg Charitable Trust et le Dr. Dvora and Haim Teitelbaum Endowment Fund.
Le professeur Hornstein est le titulaire de la chaire professorale de la famille Mondry.